
EBI-Metagenomics/envident main @ 269afbd
 View on GitHub
View on GitHub
![]()
Introduction
EBI-Metagenomics/envident EBI-Metagenomic's eDNA analysis pipeline. This pipeline is designed for the analysis of environmental DNA (eDNA) sequencing data, implementing a comprehensive workflow for quality control, primer identification, Amplicon Sequence Variant (ASV) calling and taxonomic profiling using modern bioinformatics tools. Currently the pipeline supports analysis of COI metabarcoding reads.
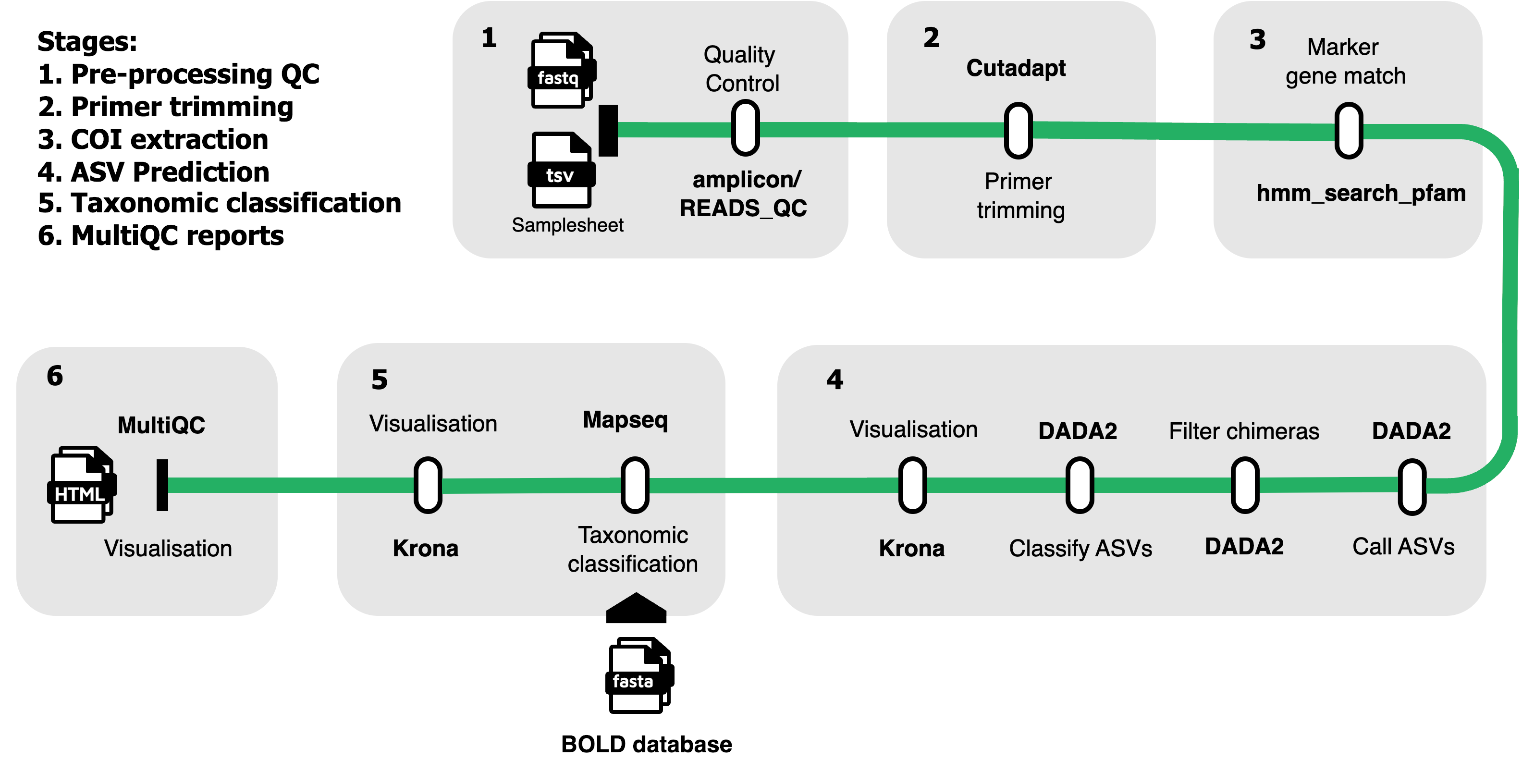
Default steps in EnvIdent
Quality Control and Preprocessing:
- Raw reads quality assessment using FastQC
- Reads quality control and filtering using fastp
- Minimum read count filtering (configurable threshold)
Primer Analysis:
- Automatic primer identification using PIMENTO
- Primer trimming using Cutadapt
- Primer validation and reporting
Taxonomic Profiling:
- Pfam-based COI (Cytochrome C Oxidase subunit I) profiling using HMMER
- Reads percentage threshold filtering for marker gene identification (configurable threshold)
ASV Analysis:
- Amplicon Sequence Variant (ASV) calling using DADA2
- ASV taxonomic classification using MAPseq
- Krona chart visualization for taxonomic results
Reporting and Quality Control:
- Comprehensive MultiQC reports
- Failed and passed runs tracking
- Software version reporting
Usage
[!NOTE] If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with
-profile testbefore running the workflow on actual data.
Requirements
The pipeline requires:
- Nextflow (≥24.04.2)
- Docker, Singularity, or Conda for software management
- Access to reference databases
- Primer database formatted for PIMENTO - a FASTA file with contig ids ending with F for forward primers and R for reverse primers. See here for an example
Input Format
The input data should be eDNA sequencing reads (paired-end or single-end) in FASTQ format, specified using a CSV samplesheet:
sample,fastq_1,fastq_2,single_end
sample1,/path/to/sample1_R1.fastq.gz,/path/to/sample1_R2.fastq.gz,false
sample2,/path/to/sample2.fastq.gz,,true
Basic execution
nextflow run EBI-Metagenomics/envident \
-r main \
-profile example_slurm \
--input samplesheet.csv \
--outdir results
Pipeline output
Example output structure for a sample (sample1). The qc_passed and qc_failed csvs are only present if you have samples that passed or failed:
results/
├── sample1/
│ ├── asv/
│ │ ├── sample1_DADA2-BOLD_asv_read_counts.tsv
│ │ ├── sample1_DADA2-MIDORI_asv_read_counts.tsv
│ │ └── sample1_dada2_stats.tsv
│ │ └── sample1_asvs.fasta
│ ├── hmmsearch-COI/
│ │ ├── sample1_Pfam-A.domtbl
│ │ └── sample1_Pfam-A.txt
│ ├── primer-identification/
│ │ └── sample1.cutadapt.json
│ ├── qc/
│ │ ├── sample1_seqfu.tsv
│ │ └── sample1.fastp.json
│ │ └── sample1.merged.fastq.gz
│ │ └── sample1_suffix_header_err.json
│ ├── taxonomy-summary/
│ │ ├── DADA2-BOLD/
│ │ | ├── ERR8441464_DADA2-BOLD_asv_krona_counts.txt
│ │ | ├── ERR8441464_DADA2-BOLD_asv_taxa.tsv
│ │ | ├── ERR8441464_DADA2-BOLD.html
│ │ | └── ERR8441464_DADA2-BOLD.mseq
│ │ ├── DADA2-MIDORI/
│ │ | ├── ERR8441464_DADA2-MIDORI_asv_krona_counts.txt
│ │ | ├── ERR8441464_DADA2-MIDORI_asv_taxa.tsv
│ │ | ├── ERR8441464_DADA2-MIDORI.html
│ │ | └── ERR8441464_DADA2-MIDORI.mseq
├── pipeline_info/
│ ├── execution_report_YYYY-MM-DD_HH-mm-ss.html
│ ├── execution_timeline_YYYY-MM-DD_HH-mm-ss.html
│ ├── execution_trace_YYYY-MM-DD_HH-mm-ss.txt
│ ├── params_YYYY-MM-DD_HH-mm-ss.json
│ ├── pipeline_dag_YYYY-MM-DD_HH-mm-ss.html
│ └── envident_software_mqc_versions.yml
├── multiqc_report.html
├── qc_passed_runs.csv
└── qc_failed_runs.csv
Credits
EBI-Metagenomics/envident was written by Christina Vasilopoulou and Jennifer Mattock.
Citations
This pipeline uses code developed and maintained by the nf-core community, reused here under the MIT license.
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
Please cite this pipeline using the following DOI: 10.48546/workflowhub.workflow.2177.1
Version History
main @ 269afbd (earliest) Created 26th May 2026 at 18:04 by Jennifer Mattock
Merge pull request #4 from EBI-Metagenomics/readme-update
Updated README.md, added example slurm config file
Frozen
 main
main269afbd
 Creators and Submitter
Creators and SubmitterViews: 684 Downloads: 323
Created: 26th May 2026 at 18:04
Last updated: 26th May 2026 at 18:17
TagsThis item has not yet been tagged.
AttributionsNone
Related items

MGnify (formerly known as EBI Metagenomics) is a free resource for the assembly, analysis, archiving and browsing all types of microbiome derived sequence data.
Space: This Team is not associated with a Space
Public web page: https://www.ebi.ac.uk/metagenomics/