
OpenEBench TCGA Cancer Driver Genes benchmarking workflow
 View on GitHub
View on GitHubDescription
The workflow takes an input file with Cancer Driver Genes predictions (i.e. the results provided by a participant), computes a set of metrics, and compares them against the data currently stored in OpenEBench within the TCGA community. Two assessment metrics are provided for that predicitions. Also, some plots (which are optional) that allow to visualize the performance of the tool are generated. The workflow consists in three standard steps, defined by OpenEBench. The tools needed to run these steps must be in one or more Docker images, generated from Docker containers. Separated instances are spawned from these images for each step:
- Validation: the input file format is checked and, if required, the content of the file is validated (e.g check whether the submitted gene IDs exist)
- Metrics Computation: the predictions are compared with the 'Gold Standards' provided by the community, which results in two performance metrics - precision (Positive Predictive Value) and recall(True Positive Rate).
- Results Consolidation: the benchmark itself is performed by merging the tool metrics with the rest of TCGA data. The results are provided in JSON format and SVG format (scatter plot).
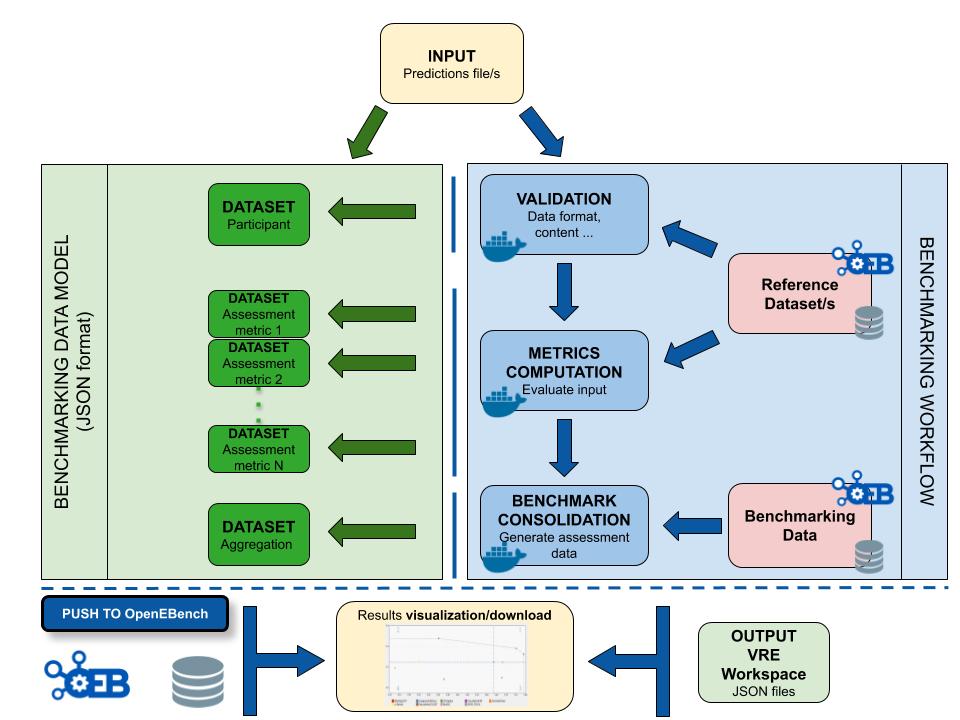
Data
- TCGA_sample_data folder contains all the reference data required by the steps. It is derived from the manuscript:
Comprehensive Characterization of Cancer Driver Genes and Mutations, Bailey et al, 2018, Cell

- TCGA_sample_out folder contains an example output for a worklow run, with two cancer types / challenges selected (ACC, BRCA). Results obtained from the default execution should be similar to those ones available in this directory. Results found in TCGA_sample_out/results can be visualized in the browser using
benchmarking_workflows_results_visualizerjavascript library.
Usage
In order to use the workflow you need to:
- Install Nextflow, which depends on Java virtual machine (>=8 , <15 ). You can automate their installation for local testing using run_local_nextflow.bash.
- Clone TCGA benchmarking Docker definitions repository from tag 1.0.3 in a separate directory, and build locally the three Docker images found in it, running the
build.sh 1.0.3script within that repo. - Run it just using either
nextflow run main.nf -profile dockeror./run_local_nextflow.bash run main.nf -profile docker. Arguments specifications:
Usage:
Run the pipeline with default parameters:
nextflow run main.nf -profile docker
Run with user parameters:
nextflow run main.nf -profile docker --predictionsFile {driver.genes.file} --public_ref_dir {validation.reference.file} --participant_name {tool.name} --metrics_ref_dir {gold.standards.dir} --cancer_types {analyzed.cancer.types} --assess_dir {benchmark.data.dir} --results_dir {output.dir}
Mandatory arguments:
--input List of cancer genes prediction
--community_id Name or OEB permanent ID for the benchmarking community
--public_ref_dir Directory with list of cancer genes used to validate the predictions
--participant_id Name of the tool used for prediction
--goldstandard_dir Dir that contains metrics reference datasets for all cancer types
--event_id List of types of cancer selected by the user, separated by spaces
--assess_dir Dir where the data for the benchmark are stored
Other options:
--validation_result The output file where the results from validation step will be saved
--assessment_results The output file where the results from the computed metrics step will be saved
--aggregation_results The output file where the consolidation of the benchmark will be saved
--statistics_results The output directory with nextflow statistics
--outdir The output directory where the consolidation of the benchmark will be saved
--statsdir The output directory with nextflow statistics
--data_model_export_dir The output dir where json file with benchmarking data model contents will be saved
--otherdir The output directory where custom results will be saved (no directory inside)
Flags:
--help Display this message
Default input parameters and Docker images to use for each step can be specified in the config file NOTE: In order to make your workflow compatible with the OpenEBench VRE Nextflow Executor, please make sure to use the same parameter names in your workflow.
Version History
Version 4 (latest) Created 29th Nov 2021 at 15:21 by Laura Rodriguez-Navas
updated repository tag from 1.0.7 to 1.0.8
Open
 master
masterf6d3a16
Version 3 Created 25th Nov 2021 at 13:31 by Laura Rodriguez-Navas
updated repository tag from 1.0.6 to 1.0.7
Frozen
master
e17c7bd
Version 2 Created 24th Nov 2021 at 14:03 by Laura Rodriguez-Navas
updated repository tag from 1.0.4 to 1.0.6
Frozen
master
0bbd457
Version 1 (earliest) Created 23rd Nov 2021 at 16:55 by Laura Rodriguez-Navas
Added/updated 1 files
Frozen
master
3a13322
 Creators and Submitter
Creators and SubmitterCreators


Additional credit
Javier Garrayo-Ventas
Submitter

Views: 8286 Downloads: 2843
Created: 23rd Nov 2021 at 16:55
Last updated: 29th Nov 2021 at 15:41
Tags AttributionsNone
Related items


Teams: OpenEBench
Organizations: Barcelona Supercomputing Center
 https://orcid.org/0000-0002-4159-6096
https://orcid.org/0000-0002-4159-6096

Teams: GalaxyProject SARS-CoV-2, nf-core viralrecon, EOSC-Life - Demonstrator 7: Rare Diseases, iPC: individualizedPaediatricCure, EJPRD WP13 case-studies workflows, TransBioNet, OpenEBench, ELIXIR Proteomics
Organizations: Barcelona Supercomputing Center, ELIXIR
https://orcid.org/0000-0003-4929-1219
Computer Engineer in Barcelona Supercomputing Center (BSC)

A space managed by WorkflowHub administrators for teams that don't want/need to manage their own space.
Teams: IBISBA Workflows, NMR Workflow, UNLOCK, NanoGalaxy, Galaxy Climate, PNDB, IMBforge, COVID-19 PubSeq: Public SARS-CoV-2 Sequence Resource, LBI-RUD, Nick-test-team, Italy-Covid-data-Portal, UX trial team, Integrated and Urban Plant Pathology Laboratory, SARS-CoV-2 Data Hubs, lmjxteam2, virAnnot pipeline, Ay Lab, iPC: individualizedPaediatricCure, Harkany Lab, MOLGENIS, EJPRD WP13 case-studies workflows, Common Workflow Language (CWL) community, Testing, SeBiMER, IAA-CSIC, MAB - ATGC, Probabilistic graphical models, GenX, Snakemake-Workflows, ODA, IPK BIT, CO2MICS Lab, FAME, CHU Limoges - UF9481 Bioinformatique / CNR Herpesvirus, Quadram Institute Bioscience - Bioinformatics, HecatombDevelopment, Institute of Human Genetics, Testing RO Crates, Test Team, Applied Computational Biology at IEG/HMGU, INFRAFRONTIER workflows, OME, TransBioNet, OpenEBench, Bioinformatics and Biostatistics (BIO2 ) Core, VIB Bioinformatics Core, CRC Cohort, ICAN, MustafaVoh, Single Cell Unit, CO-Graph, emo-bon, TestEMBL-EBIOntology, CINECA, Toxicology community, Pitagora-Network, Workflows Australia, Medizinisches Proteom-Center, Medical Bioinformatics, AGRF BIO, EU-Openscreen, X-omics, ELIXIR Belgium, URGI, Size Inc, GA-VirReport Team, The Boucher Lab, Air Quality Prediction, pyiron, CAPSID, Edinburgh Genomics, Defragmentation TS, NBIS, Phytoplankton Analysis, Seq4AMR, Workflow registry test, Read2Map, SKM3, ParslRNA-Seq: an efficient and scalable RNAseq analysis workflow for studies of differentiated gene expression, de.NBI Cloud, Meta-NanoSim, ILVO Plant Health, EMERGEN-BIOINFO, KircherLab, Apis-wings, BCCM_ULC, Dessimoz Lab, TRON gGmbH, GEMS at MLZ, Computational Science at HZDR, Big data in biomedicine, TRE-FX, MISTIC, Guigó lab, Statistical genetics, Delineating Regions-of-interest for Mass Spectrometry Imaging by Multimodally Corroborated Spatial Segmentation, OLCF-WES, Bioinformatics Unit @ CRG, Bioinformatics Innovation Lab, BSC-CES, ELIXIR Proteomics, Black Ochre Data Labs, Zavolan Lab, Metabolomics-Reproducibility, Team Cardio, NGFF Tools, Bioinformatics workflows for life science, Workflows for geographic science, Pacific-deep-sea-sponges-microbiome, CSFG, SNAKE, Katdetectr, INFRAFRONTIER GmbH, PerMedCoE, Euro-BioImaging, EOSC-Life WP3 OC Team, cross RI project, ANSES-Ploufragan, SANBI Pathogen Bioinformatics, Biodata Analysis Group, DeSci Labs, Erasmus MC - Viroscience Bioinformatics, ARA-dev, Mendel Centre for Plant Genomics and Proteomics, Metagenomic tools, WorkflowEng, Polygenic Score Catalog, bpm, scNTImpute, Systems Biotechnology laboratory, Cimorgh IT solutions, MLme: Machine Learning Made Easy, Hurwitz Lab, Dioscuri TDA, Scipion CNB, System Biotechnology laboratory, yPublish - Bioinfo tools, NIH CFDE Playbook Workflow Partnership, MMV-Lab, EMBL-Bioimage Analysis Support, EBP-Nor, Evaluation of Swin Transformer and knowledge transfer for denoising of super-resolution structured illumination microscopy data, Bioinformatics Laboratory for Genomics and Biodiversity (LBGB), multi-analysis dFC, CholGen, RNA group, Plant Genomes Pipelines in Galaxy, Pathogen Genomic Laboratory, Chemical Data Lab, JiangLab, Pangenome database project, HP2NET - Framework for construction of phylogenetic networks on High Performance Computing (HPC) environment, Center for Open Bioimage Analysis, Generalized Open-Source Workflows for Atomistic Molecular Dynamics Simulations of Viral Helicases, Historical DNA genome skimming, QCDIS, Peter Menzel's Team, NHM Clark group, ESRF Workflow System (Ewoks), Kalbe Bioinformatics, Nextflow4Metabolomics, GBCS, CEMCOF, Jackson Laboratory NGS-Ops, Schwartz Lab, BRAIN - Biomedical Research on Adult Intracranial Neoplasms, Cancer Therapeutics and Drug Safety, Deepdefense, Mid-Ohio Regional Planning Commission, MGSSB, Institute for Human Genetics and Genomic Medicine Aachen, FengTaoSMU, EGA, Plant-Food-Research-Open, KrauthammerLab, Geo Workflows, grassland pDT, FunGIALab, CRIM - Computer Research Institute of Montréal, Medvedeva Lab, Metagenlab, FAIR-EASE, Protein-protein and protein-nucleic acid binding site prediction research, Culhane Lab, IDUN - Drug Delivery and Sensing, Edge Computing DAG Task Scheduling Research Group, Stratum corneum nanotexture feature detection using deep learning and spatial analysis: a non-invasive tool for skin barrier assessment, COPO, Taudière group, ErasmusMC Clinical Bioinformatics, interTwin, fluid flow modeling, EnrichDO, WorkflowResearch, Application Security - Test Crypt4GH solutions, RenLabBioinformatics, Yongxin's team, PiFlow, HLee_SeoGroup, UFZ - Image Data Management and Processing Workflows, Korean Bioinformaticians, Into the deep, XChem, CPM, SocialGene, Research Data Management ICE-2, ObjectRecognition, LiDAR, FONDA II C2, Astroparticle Lab, FAIRagro M4.4, Kgerring, QuackenbushLab, Virus sequencing team, SOS, BioImage Informatics and Analysis Workflows, BoostNano, simblockflow, CSSB, Research on Workflow scheduling, Research Data1, CSUbioinformatics, CDPP, Mr., ASD-HRS, data management, FAIR_thesis: Marine acoustic data, CellBinDB, DEEP Lab, University of Amsterdam, SIMEXP, nf-pediatric Team, Kasmanas, Structural Variation Analysis, fuzzyworkflow, CausalCoxMGM Team, Tufts University Center for Cellular Agriculture (TUCCA), Test, CrustyBase, Applied Computational Cancer Research, Click-qPCR, BAID Team, FabianLab, Vector informatics and genomics group, AlmondBreedingLab, Artificial Design for Intelligent Breeding, ELIXIR Biodiversity Community, GROTIA, Biomedical_LLM, WhiteSymmetry, Hämatologie Labor Kiel, pakbaba, Metagenomics Analysis, MTB Bioinformatics Workflows, RTC Bioinformatics, CMG-GUTS, EI Papatheodorou Group, Metagenomics Analysis of Microbiome, ColoPola: A polarimetric imaging dataset for colorectal cancer detection, AI in Zhou Lab, CEPLAS, LTER-Italy, Gevaert Lab, Kendall-Theisen lab pipelines, High Performance Scientific Computing Laboratory (HPSC), Ensembl Metazoa and Plants, BiRD, Q-Dawn, AMRMALDI, Systems and Synthetic Biology, EI Core Bioinformatics Group, Values Shape Community, SolutionMake, GENEX, iPol, zxfenglab, Bioinformatics Unit IIS-FJD, omnibenchmark, BioX Fanatics, Li-Omics Lab, Metro Team, ZiemertLab, Song Lab, PanGIA, Holobionts Workflows, An Ideal Design for RNA-seq investigations into RPL, AMI2B Team, Institute for Hearing Technology and Acoustics (IHTA), Transcriptomics in unexplained recurrent pregnancy loss, E-MOBI / EKONOMIK MOBIL,S.R.L, Workflow Run Crate working group, datafun, BioFAIR, BH2025_Project30, NII Test Team, LIB, Galaxy and Beyond, Biodepot Spatial Proteomics Team, WEHI SODA Hub, Digital Platform, Anesthesia and Neurocritical Care Medicine, Digital Skills in Arts and Humanities, BioimageAIpub, Dalmolin Systems Biology Group, FAIRMD, KG-Microbe, BGIQD, CSB, EMRS, BioImagingUK, Bachelor Thesis Developeming an Algorithm for the Generation of Synthetic Scientific Workflows and Characterization of the generated Workflows, GenotoulBioinfo, mzlab, Liver Polyploidy Research Team, Computational Metabolomics (UoB), UiT-CANS, PathoFact2, Polyploid Spatial Transcriptomics Team, CCBR, FONDA T9 Project Team, Cen Occitanie's Geomateam, Exeter Genetics, HP-Phasing, CPSM, Bayrak Lab, Eco-Flow, oceanbase, Proj Renzo, UFZ Galaxy workflows, CEIT-Metaverse, EnChemTox, Tesla, FHAIVE - Finnish Hub for Development and Validation of Integrated Approaches, ZhuLab MII Group, Protea Glycosciences Analytical, Immunodynamics-Engel-Lab, NanoporeDB, SyBR Development, Membrane Protein-Lipid Interaction Database Pipeline, Grunwald Lab, SensorWF, Chocot test team, UNSW MWAC Structural Biology Facility, ELIXIR Norway, EMBL Hentze Group, ConesaLab
Web page: Not specified

OpenEBench (https://openebench.bsc.es) is the ELIXIR benchmarking and technical monitoring platform for bioinformatics tools, web servers, and workflows. OpenEBench is part of the ELIXIR Tools platform and its development is led by the Barcelona Supercomputing Center (BSC) in collaboration with partners within ELIXIR and beyond.
Within the ELIXIR project, OpenEBench is being developed under the Tools Platform at the Work Package 2 (WP2: Benchmarking) (https://elixir-europe.org/platforms/tools). ...
Space: Independent Teams
Public web page: https://openebench.bsc.es